
Structural Genomics: The Repetitive Component of Plant Genomes
Transposons are a major component of eukaryotic genomes
The genome is made of DNA of a cell. The genome size in eukaryotes is very different; in general, this variation is not related to the information content, i.e. the number of genes.

Genes (encoding proteins) represent only a small portion of the genome. Much of the genome consists of repeated sequences that have increased their presence in the course of evolution.
The variability of the genome size is mainly related to variations in the frequency of repeated, “non-coding”, sequences, especially transposons. Transposons (or transposable elements, TEs) are sequences of DNA that “transpose” themselves from one site to another in the genome. They represent the vast majority of repeated sequences in eukaryotic genomes.
TEs are subdivided into two main classes accordingly to their mechanism of transposition, retrotransposons (REs, class I) and DNA transposons (class II).
Class I elements, which includes all REs, can transpose through a replicative mechanism similar to that of retrovirus, which involves the transcription of an RNA intermediate by the enzyme machinery of the host cell, and subsequent retrotranscription to cDNA and integration into the host genome by the enzymes encoded by the retrotransposon RNA.
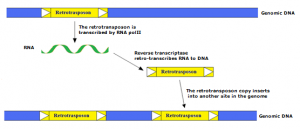
Such a “copy and paste” mechanism has been largely successful during the evolution of eukaryotes in which class I elements represent the largest portion of higher plant genomes.
Function of retrotransposons
Over the last two decades, some examples have correlated the emerging of RE activity in the genome with a stress mediated reaction: Tnt1 and Tto1 in Nicotiana and Tos17 in rice showed stress-induced (by tissue culture) transcription and transposition, while these elements were not transcribed in standard culture conditions.
Large genome sequencing of grass plants showed that REs are responsible for extensive changes in genome structure and, surprisingly, dramatic differences were reported even among individuals belonging to the same species.
It has been proposed that REs restructuring action plays a role in regulating gene expression. It has been suggested that allelic variation in non-genic (regulatory) sequence may be involved in heterosis, i.e. the superior performance of hybrids in respect of their parents. In this sense, the old epithet of “junk” for such repeated sequences, which have affected genome structure and function, is becoming obsolete.
Present work at the Genetics and Genomics Lab

Sunflower (Helianthus annuus L.)
- Mascagni et al. Specific LTR-Retrotransposons Show Copy Number Variations between Wild and Cultivated Sunflowers. Genes 9, 433, 2018
- Mascagni et al. Repetitive DNA and Plant Domestication: Variation in Copy Number and Proximity to Genes of LTR-Retrotransposons among Wild and Cultivated Sunflower (Helianthus annuus) Genotypes. Genome Biology and Evolution 7, 3368–3382, 2015.
The relationship between variation of the repetitive component of the genome and domestication in plant species is not fully understood. The sunflower (Helianthus annuus) genome contains a very large proportion of transposable elements, especially long terminal repeat retrotransposons. We used NGS technologies to perform a quantitative and qualitative survey of intraspecific variation of the retrotransposon fraction of the genome across 15 genotypes—7 wild accessions and 8 cultivars—of H. annuus. By mapping the Illumina reads of the 15 genotypes onto a library of sunflower long terminal repeat retrotransposons, we observed considerable variability in redundancy among genotypes, at both superfamily and family levels. Large variability among genotypes was also ascertained for retrotransposon proximity to genes. Both redundancy and proximity to genes varied among retrotransposon families and also between cultivated and wild genotypes. These data suggest that structural variations related to retrotransposons might have occurred to produce phenotypic variation between wild and domesticated genotypes.

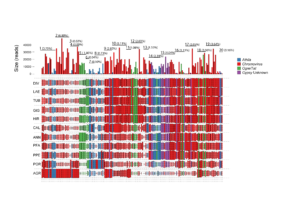
Redundancy of Gypsy and Copia superfamilies (A) and families (B) in 15 sunflower genotypes. Bars not sharing the same letter are to be considered as different according to a threshold indicating the extent of differences related to random sampling of reads. The respective genome proportion of the superfamily or family is reported inside brackets. Within each bar, state codes indicate wild North-American accessions, names indicate cultivars.
Mascagni F. et al. Genome-wide analysis of LTR retrotransposon diversity and its impact on the evolution of the genus Helianthus (L.). BMC Genomics 18, 634, 2017
Genome divergence by mobile elements activity and recombination is a continuous process that plays a key role in the evolution of species. Nevertheless, knowledge on retrotransposon-related variability among species belonging to the same genus is still limited. Considering the importance of the genus Helianthus, a model system for studying the ecological genetics of speciation and adaptation, we performed a comparative analysis of the repetitive genome fraction across ten species and one subspecies of sunflower, focusing on long terminal repeat retrotransposons at superfamily, lineage and sublineage levels. On average, repetitive DNA in Helianthus species represented more than 75% of the genome, being composed mostly by long terminal repeat retrotransposons. We found considerable variability in the abundance of diverse retrotransposon lineages and sublineages. This large variability should indicate that different events of amplification or loss related to these elements occurred following species separation and should have been involved in species differentiation. Moreover, the data suggested that LTR-RE abundance in a species was affected by the annual or perennial habit of that species.
![]()
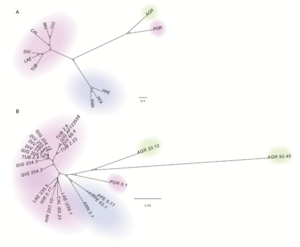
(above) Overall picture of the LTR-Gypsy-RE-related families. The size of the rectangle is proportional to the genome proportion of a family for each species (on the right). Bar plot in the top row shows the size of the families.The colour of the rectangles corresponds to the lineage of the Gypsy LTR-RE; (below) A Dendrogram obtained by a hierarchical clustering analysis based on genome proportion data of Copia- and Gypsy-related families of different Helianthus species. The bar represents the genetic distance. B Phylogram of the same Helianthus species based on ETS sequences. Colours indicate the different analysed botanical sections: pink for Divaricati, light blue for Helianthus and green for Agrestis section.

Olive (Olea europaea L.)
Barghini E. et al. LTR retrotransposon dynamics in the evolution of the olive (Olea europaea) genome. DNA Research 22, 91-100, 2015.
In this study, we provided characterization of a sample of full-length long terminal repeat (LTR)
REs in the olive genome. Mapping a large set of Illumina whole-genome shotgun reads onto the identified retroelement set revealed that Gypsy elements are more redundant than Copia elements. The insertion time of intact retroelements was estimated based on sister LTR’s divergence. Although some elements inserted relatively recently, the mean insertion age of the isolated retroelements is around 18 million yrs. Gypsy and Copia retroelements showed different waves of transposition, with Gypsy elements especially active between 10 and 25 million yrs ago and nearly inactive in the last 7 million yrs. The occurrence of numerous solo-LTRs related to isolated full-length retroelements was ascertained for two Gypsy elements and one Copia element. Overall, the results reported in this study show that RE activity (both retrotransposition and DNA loss) has impacted the olive genome structure in more ancient times than in other angiosperms.
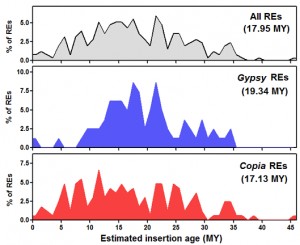
Distributions of full-length REs identified in this study, according to their estimated insertion ages (MY). Mean insertion dates are reported in parentheses.
Barghini E. et al. Identification and characterisation of Short Interspersed Nuclear Elements in the olive tree (Olea europaea L.) genome. Molecular Genetics and Genomics 292, 53-61, 2017.
Short Interspersed Nuclear Elements (SINEs) are non autonomous retrotransposons in the genome of most eukaryotic species. While SINEs have been intensively investigated in humans and other animal systems, SINE identification has been carried out only in a limited number of plant species. The aim of this work was to produce a specific bioinformatics pipeline for analysing second generation sequence reads and identifying SINEs. We have identified, for the first time, 227 putative SINEs of the olive tree, that constitute one of the few sets of such sequences in dicotyledonous species. A comparison of sequence conservation between olive SINEs and LTR retrotransposon families suggested that SINE expansion in the genome occurred especially in very ancient times, before LTR retrotransposon expansion, and presumably before the separation of the rosids (to which Oleaceae belong) from the Asterids.
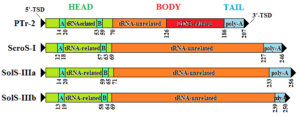
Schematic representation of the major SINE types identified in the olive genome. The head, the body, and the tail of the SINE are indicated. Numbers indicate the average coordinates of each region (A, B A- and B- box, respectively, as indicated by SINESearch tool; TSD target site duplication).

Poplars (Populus spp.)
- Usai et al. Comparative genome-wide analysis of repetitive DNA in the genus Populus L.. Tree Genetics & Genomes, 13, 96, 2017.
Genome skimming was performed, using Illumina sequence reads, in order to obtain a detailed comparative picture of the repetitive component of the genome of Populus species. The repetitive portion of the genome ranged from 33.8 in Populus nigra to 46.5% in Populus tremuloides. The large majority of repetitive sequences were long terminal repeat-retrotransposons. Gypsy elements were over-represented compared to Copia ones, with a mean ratio Gypsy to Copia of 6.7:1. Satellite DNAs showed a mean genome proportion of 2.2%. DNA transposons and ribosomal DNA showed genome proportions of 1.8 and 1.9%, respectively. Concerning Copia lineages, similar transpositional profiles were observed among all the analysed species; by contrast, differences in transpositional peaks of Gypsy lineages were found. Overall, the data indicate that the repetitive component of the genome in the poplar genus is still rapidly evolving.
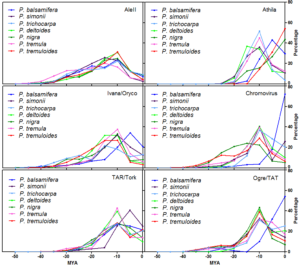
Timing of retrotranspositional activity of six LTR-RE lineages in seven poplar species, based on the pairwise comparisons of Illumina reads matching RT-encoding sequences. The y axis reports the product of the percentage of pairwise comparisons for the average coverage of the RT sequence in each species.

Other Research Lines
Ficus carica genome
Ficus carica L. is a diploid species, with a genome size of 0.36 pg/2C. With the aim of analysing the fig genome structure, we used Illumina technology to produce around 40 genome equivalents of sequence reads which were assembled into contigs and scaffolded. This first assembly is composed of 264,088 scaffolds, up to 41,760 nt in length, covering 323,708,138 nt, that corresponds to 87.5% of the fig genome, with N50 = 2,523. Coding genes account for at least 6.8% of the fig genome. Concerning the repetitive component, the fig genome is composed for around 58% of repeated sequences, of which none was especially redundant. Among identified repeats, the most represented are LTR-retrotransposons.

applications of next generation sequencing to the genomics of Posidonia oceanica.
Posidonia oceanica is a monocotyledonous marine plant that plays a crucial role in maintaining the Mediterranean environment. Despite its ecological importance, basic knowledge of the functional and structural genomics of this species is still limited, as it is for the other seagrasses. We are studying, for the first time, the genome structure of this seagrass using a low coverage of Illumina sequences and different assembly approaches. A very large proportion of the genome is represented by long-terminal-repeat (LTR) retrotransposons of both the Copia and Gypsy superfamilies. Posidonia LTR retrotransposons are classified and their sequences analysed. Analysis of sequence variability indicate that Gypsy families have experienced amplification in more recent times compared to Copia ones.
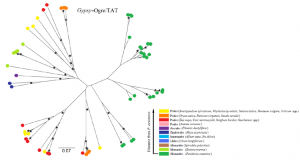
Phylogenetic tree of a Gypsy family of retroelements of Posidonia oceanica and other monocotyledons. For each sequence, the species or the group of species is indicated by the color reported in the bottom, in which the relative distance of each species or group of species from P. oceanica is reported. Scale bars indicate the distance. Asterisks indicate bootstrap values higher than 50%.