
Functional Genomics: Analysis of Gene Expression using RNAseq
NGS analyses of transcriptomes
The recent development of high-throughput sequencing technologies are changing the way transcriptomes are discovered and defined. For example, the Illumina system can yield millions of short reads and is therefore more suitable for tag-based transcriptome sequencing and digital gene expression analysis, achieving a superior accuracy and precision compared to older methods, especially for genes showing low levels of expression.
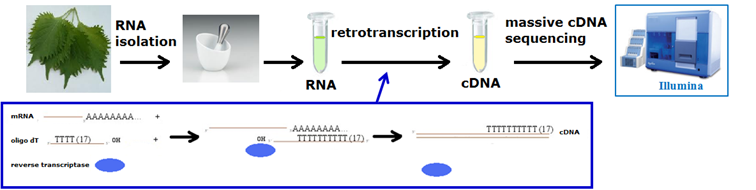
It is possible to sequence the whole RNA produced by a tissue (or a cell) and then analyze gene expression with extreme precision. Once obtained the sequence of the whole RNA produced by the tissue, in case the transcriptome of that species is not available, sequence reads can be assembled, reconstructing complete cDNAs, and annotated. When the complete transcriptome of the species has been obtained or if it is already available, the level of gene expression is measured “by counting” sequences complementary to the gene sequences (mapping).
The real challenge is represented by managing billions of sequence data and deducing metabolic pathways and understanding the biological meaning of changes in gene expression.

A. Vangelisti et al. How an ancient, salt-tolerant fruit crop, Ficus carica L., copes with salinity: a transcriptome analysis. Scientific Reports, in press (2019)
Although Ficus carica L. (fig) is one of the most resistant fruit tree species to salinity, no comprehensive studies are currently available on its molecular responses to salinity. Here we report a transcriptome analysis of F. carica cv. Dottato exposed to 100 mM sodium chloride for 7 weeks, where RNA-seq analysis was performed on leaf samples at 24 and 48 days after the beginning of salinization; a genome-derived fig transcriptome was used as a reference. At day 24, 224 transcripts were significantly up-regulated and 585 were down-regulated, while at day 48, 409 genes were activated and 285 genes were repressed. Relatively small transcriptome changes were observed after 24 days of salt treatment, showing that fig plants initially tolerate salt stress. However, after an early down-regulation of some cell functions, major transcriptome changes were observed after 48 days of salinity. Seven weeks of 100 mM NaCl dramatically changed the repertoire of expressed genes, leading to activation or reactivation of many cell functions. We also identified salt-regulated genes, some of which had not been previously reported to be involved in plant salinity responses. These genes could be potential targets for the selection of favourable genotypes, through breeding or biotechnology, to improve salt tolerance in fig or other crops.


A. Vangelisti et al. Transcriptome changes induced by arbuscular mycorrhizal fungi in sunflower (Helianthus annuus L.) roots. Scientific Reports 8, 4, 2018.
Mycorrhization concerns mutualistic symbiosis between more than two-thirds of all known plant species, including important crop species, and fungi of the Glomeromycota phylum (arbuscular mycorrhizae). Fungal extra-radical hyphae absorb from the soil mineral nutrients, mainly phosphorus (P) and nitrogen (N), which are available for the host plant. At the same time the plant supplies fungus with sugars generated by photosynthesis. At molecular level this process involves many molecular signals and gene products of both plant and fungus but in sunflower these are poorly studied and transcriptomic analyses through NGS allowed us to identify new unknown factors involved in this process.

T. Giordani et al. Genome-wide analysis of LTR-retrotransposon expression in leaves of Populus × canadensis water-deprived plants. Tree Genetics & Genomes 12, 75, 2016.
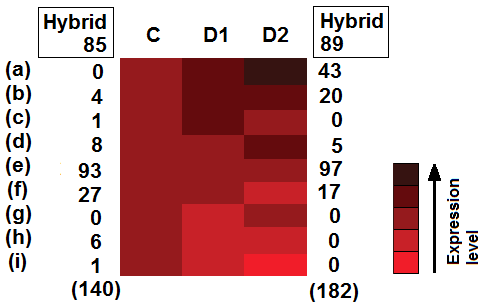
Retrotransposons represent a major component of plant genomes; however, large-scale studies on their expression are rare. We evaluated the expression of long terminal repeat-retrotransposons in leaves of two sister hybrids Populus × canadensis (P. deltoides × P. nigra), subjected to moderate or severe water deprivation by mapping Illumina RNA-Seq reads onto a set of 958 unique full-length retrotransposons of P. trichocarpa. Detectable levels of transcription were ascertained for 140 retrotransposons in 1 hybrid and 182 in the other. The number of expressed Gypsy elements in control and water-deprived plants was higher than those of Copia, as were their expression levels. The two hybrids showed different retrotransposon expression patterns following water deprivation. Such variations between hybrids were related to differential expression of a few genes involved in chromatin methylation and remodeling.

Transcriptome analyses on milk produced by sheep supplemented with highly valuable nutraceutical compounds (in collaboration with DAFE Zootechnics Lab)
It is known that milk nutrient composition depends on food quality and food availability. Also milk nutraceuticals compounds (products derived from food sources that are purported to provide extra health benefits) can change with animal diet. For example, dairy ewes feeding with linseed or oil increases the amount of conjugated linoleic acids (CLA) in milk and this result in dairy products rich of poly-unsaturated fatty acids. It has been observed that CLA give a high lipid oxidative stability in dairy products. Moreover CLA are nutraceutical compounds able to protect from several diseases. Nothing is known on the activation of genes involved in milk enrichment. Our aim is to study gene expression changes in milk somatic cells in order to understand molecular mechanisms induced by potential nutraceutical compounds (linseed, olive polyphenols), that lead changes in milk composition.